Stem Cell Therapy for Brain Tumors
Received 3 September 2015; Accepted 24 September 2015; Publication 30 October 2015
Alexander Aleynik and Pranela Rameshwar*
- Department of Medicine - Division of Hematology/Oncology, New Jersey Medical School, Rutgers Biomedical and Health Sciences, Newark, NJ 07103 USA*
- Corresponding Author: rameshwa@njms.rutgers.edu
Abstract
Glioblastoma multiforme (GBM), the most common and lethal brain cancer, prognosis remains bleak with a median survival of about 15 months despite maximal surgical resection, radiotherapy, and temozolomide treatment. The difficulty associated with safely and effectively delivering therapeutics across the blood brain barrier (BBB) is a major challenge towards GBM treatment. Ongoing research and clinical trials, including attempts to deliver therapeutics within stem cells present possible solutions. The relationships between brain cancer pathology, stem cell properties, therapeutic advantages and disadvantages of various stem cell types, drug delivery methods, cancer stem cells, gene therapy, anti-cancer vaccines, chimeric antigen receptor therapies, and combination therapies are discussed.
Abbreviations
Glioblastoma Multiforme (GBM), blood brain barrier (BBB), World Health Organization (WHO), Parkinson’s disease (PD), Alzheimer’s disease (AD), multiple sclerosis (MS), Huntington’s Disease (HD), amyotrophic lateral sclerosis (ALS) and devastating neural injuries such as traumatic brain injury (TBI) and spinal cord injury (SCI), Embryonic stem cells (ESCs), fetal stem cells (FSCs), umbilical cord blood stem cells (UCBSCs), mesenchymal stem cells (MSCs), hematopoietic stem cells (HSCs), adipose derived stem cells (ADSCs), and adult stem cells (ASCs), induced neuronal (iN), induced pluripotent stem cells (iPSCs), human induced pluripotent stem cells (hiPSCs), neural stem cells (NSCs), MSCs derived from human adipose tissue (hAM-SCs), MSCs derived from the human bone marrow (hBMSCs), umbilical cord blood-derived MSCs (UCB-MSCs), adipose-tissue-derived mesenchy-mal stem cells (AT-MSCs), breast cancer cell (BCC), central nervous system (CNS), umbilical cord blood (UCB), subventricular zone (SVZ), RNA interference (RNAi), L-dihydroxy-phenylalanine (L-DOPA), sodium-independent neutral amino acid transporter (LAT-1), iduronate 2-sulfatase enzyme (IDS), cancer stem cells (CSCs), glioma cancer stem cells (gCSCs), Marrowisolated adult multilineage inducible (MIAMI), lipid nanocapsules (LNCs), necrosis factor-related apoptosis-inducing ligand (TRAIL), death receptor 5 (DR-5), temozolomide (TMZ), mesoporous silica nanoparticles (MSNs), glioblastoma stem-like cells (GSCs), TNF-related apoptosis-inducing ligand (TRAIL), oncolytic viruses (OV), Herpes Simplex Virus (HSV), herpes simplex virus thymidine kinase (HSV-TK), Interleukin 2 (IL-2), retroviral vector and vector producing cells (RVPCs), internal ribosome entry site (IRES), internal simian virus 40 (SV40), deep vein thrombosis (DVT), pulmonary embolism (PE), HSV-TK system with an adenoviral vector (Adv-HSV-TK), Newcastle Disease Virus (NDV), Herpes Simplex Virus G207 (HSV G 207), HUJ (Hebrew University Jerusalem), BIU (Billion Infectious Units), Hebrew University Jerusalem-New Castle Disease Virus (HUJ-NDV), cancer stem cells (CSCs), Oligodendrocyte-type-2 astrocyte (O-2A), transforming growth factor-β (TGFβ), vascular endothelial growth factor (VEGF), indolamine 2,3-dioxygenase (IDO), MicroRNAs (miRNAs), Dendritic cells (DCs), antigen presenting cells (APCs), fms-like tyrosine kinase-3 ligand (Ftl3L), granulocyte-macrophage colony-stimulating factor (GM-CSF),plasmacytoid dendritic cells (PDCs), myeloid dendritic cells (MDCs), cytotoxic T lymphocyte (CTL), adoptive cell transfer (ACT), Chimeric antigen receptor (CAR), T-cell receptors (TCRs), chronic lymphocytic leukemia (CLL).
1 Introduction
Several strategies using stem cells have been tested for treating brain tumors. These include clinical trials, animal studies, and in vitro models. There are more than one hundred and twenty types of brain tumors including astrocytomas, brain stem gliomas, craniopharyngiomas, ependymomas, germ cell tumors, glioblastomas, gliomas, medulloblastomas, meningiomas, metastatic brain tumors, mixed gliomas, oligodendrogliomas, schwannomas, pinel tumors, and many others. Glioblastoma Multiforme (GBM) also called “astrocytoma, grade IV” and “glioblastoma” is the most lethal and common brain cancer [1].
Despite maximal surgical resection, radiotherapy, and temozolomide treatment median survival remains at only 14.6 months for GBM patients [2]. A major problem in the treatment of GBM is getting therapeutics across the blood brain barrier (BBB) efficiently. The delivery of therapeutics across the BBB with stem cells is a possible solution that has been tested by experimental and clinical trials. Given the paucity of effective treatments for GBM and the ability of stem cells to deliver therapeutics past the BBB and to the tumor bed, one can argue that it is worthwhile to discuss the therapeutic advantages and disadvantages of each type of stem cell as well as their experimental and clinical applications.
GBM is a heterogeneous group of diseases histologically classified as grade IV glioma by the World Health Organization (WHO) [2, 3]. On an annual basis, there are roughly 10,000 new cases of GBM in the United States and 238,000 new cases of GBM worldwide [1, 4]. GBM accounts for 15%-20% of all intracranial tumors and 50% of all brain malignancies [1, 5, 6]. The prognosis for patients with brain cancer and GBM is poor, requires life-long physician monitoring, and can be improved by novel treatment options based on the latest data.
2 Glioblastoma Multiforme Pathology
Glioblastoma Multiforme and other brain tumors have distinct pathologies that require unique treatments. GBM has been classified as primary or secondary. Primary GBM is more prevalent than secondary GBM (~90% of patients), typically occurs de novo in older patients, and is associated with genetic modifications such as EGFR over expression, PTEN (MMAC1) mutations, CDKN2A (p16) deletions, and MDM2 amplification [3, 7]. In contrast, secondary GBM typically progresses from low-grade or anaplastic astrocytomas, affects younger patients, and often contains TP53 mutations [3, 7]. Primary and secondary GBM are indistinguishable histologically; although primary GBM typically occurs in older patients and is more prevalent than secondary GBM (~90% of patients) [3, 7].
3 Sources of Stem Cells - Advantages, Disadvantages, and other Considerations
A brief synopsis of advantages and disadvantages of ESCs, MSCs, and iPSCs is summarized in Table 1. Stem cell therapy has been under consideration as a novel biological treatment option for incurable neurodegenerative diseases such Parkinson’s disease (PD), Alzheimer’s disease (AD), multiple sclerosis (MS), Huntington’s Disease (HD), amyotrophic lateral sclerosis (ALS) and devastating neural injuries such as traumatic brain injury (TBI) and spinal cord injury (SCI) [8-11].
Embryonic stem cells (ESCs), fetal stem cells (FSCs), umbilical cord blood stem cells (UCBSCs), mesenchymal stem cells (MSCs), adipose derived stem cells (ADSCs), and adult stem cells (ASCs) can differentiate into neuronal cells in vitro and have the potential to be applied for the treatment of neurological disorders including the treatment of brain tumors such as glioblastoma multiforme [11-16].
An experimental study demonstrated that the patients’ own fibroblasts can be converted into induced neuronal (iN) cells capable of expressing multiple neuron-specific proteins, generating action potentials, and forming functional synapses through the combinatorial expression of three neural lineage-specific transcription factors, Ascl1, Brn2, and Myt1l [17]. The direct formation of fibroblasts to neurons has now been termed direct reprogramming. Subsequent studies using a rat Parkinson’s Disease model showed that human fibroblasts reprogrammed with the transcriptional factors Mash1, Ngn2, Sox2, Nurr1, and Pitx3 can change human fibroblasts into DA neuron-like cells [18]. The DA neurons, which provided relief for PD symptoms, stained positive for various markers of DA neuron-like cells, exhibited DA uptake and DA production, and were electrophysiologically functional [18].
Table 1 Advantages and disadvantages of ESCs, MSCs and iPSCs for the treating neurodegenerative Disease
| Stem Cells | Advantage | Disadvantage | Refs |
|---|---|---|---|
| ESCs (Embryonic Stem Cells) | - Differentiate into all types of neural tissues |
- Ethical controversy - Spontaneous transformation - Immune rejection - Graft-induced dyskinesias (GIDs) |
65, 137 |
| MSCs (Mesenchymal Stem Cells) |
- Easy harvest - Easy expansion - No ethical controversy - Immune modulation - Immune Suppression - Lack of Immune rejection |
- Differentiation into unwanted cell types - Poor engraftment - Tumorigenic potential - Cytogenetic aberrations |
15, 30, 138-143 |
| iPSCs (Induced Pluripotent Stem Cells) |
- Abundant supply - Autologous Transplantation - No immune rejection - No ethical controversy |
- Spontaneous transformation - Transplanted cells develop pathology they are intended to treat (e.g. Lewy Bodies in cell grafts) |
19-21, 23, 25, 26, 144-147 |
Embryonic Stem Cells (ESCs) are derived from the inner cell mass of mammalian blastocysts, can grow indefinitely while maintaining pluripotency and the ability to differentiate into cells of all three germ layers [19]. Among the different types of cells that may be used in therapy, ESCs theoretically have an advantage because they can differentiate into all types of neural tissues [19].
Likewise, induced pluripotent stem cells (iPSCs), which are created through genetic manipulation, have the potential to form all types of cells including those within the neuronal and glial lineages [19]. At the moment, iPSCs can be created from a variety of somatic cell tissue sources including skin fibroblasts, blood, and hair follicles but with different efficiencies [20, 21]. Several studies have noted advancements in cell culture protocols to improve cell differentiation, cell sorting, and removal of potentially mutagenic reprogramming vectors [20-24].
While the main disadvantage of using iPSCs is their spontaneous transformation, their advantages lie in the ability to mimic the patient’s genotype, avoid immune rejection, and forego the ethical quandaries associated with using human ESCs [25]. The iPSCs have been considered as a possible treatment option for PD, ALS, MS, and AD [25]. Various combinations of genetic manipulations have been used to generate iPSCs with the ability to differentiate into various neural lineages. A recent study showed that DOX-inducible lentiviral vectors could be excised with Cre-recombinase to generate human induced pluripotent stem cells (hi PSCs) that lack potentially tumorigenic reprogramming vectors such as c-Myc [26].
Although neural stem cells (NSCs) can be generated from multiple sources such as differentiated ESCs, iPSCs, fetal tissues, and cadavers, these sources may not be able to produce adequate numbers NSCs for widespread clinical implementation. MSC therapy presents an alternative to the use of NSCs. MSCs are heterogeneous multipotent cells that can form all germ layers and which have been characterized based on several criteria previously established in literature [27, 28]. MSCs have been extracted from several adult human tissues including the connective tissues of all human organs, the bone marrow (BM), skeletal muscle, synovium, dental pulp, and adipose tissues [29-33]. MSCs can also be harvested from fetal tissues such as umbilical cord blood, placenta, amniotic membranes, and amniotic fluid [31, 33-39].
In the BM, MSCs contacting the abluminal region of the central sinus can function as “gatekeepers” that regulate BM homeostasis and function [40, 41]. In addition to the regenerative differentiation capacity of MSCs, the protective function of these cells underscores many other non-progenitor functions such as immunoenhancement, immunomodulation, and the maintenance of homeostasis as well as the maintenance of a unique niche microenvironment through the secretion of soluble factors [28, 42].
Discussions about the advantages and disadvantages of MSCs from various sources have been given considerable attention in the literature. For example, several studies have claimed that MSCs derived from human adipose tissue (hAMSCs) have similar glioma tropism to MSCs derived from the bone marrow (hBMSCs), however, hAMSCs can be harvested in larger numbers while minimizing patient morbidity during cell harvesting [43]. Another interesting study provided experimental evidence from a coculture assay with primary GBM cells showing that umbilical cord blood-derived MSCs (UCB-MSCs) inhibited GBM growth while adipose-tissue-derived MSCs (AT-MSCs) promoted GBM growth [33]. The immunosuppressive properties of MSCs can also be disadvantageous when juxtaposed with evidence showing increased breast cancer cell (BCC) growth when cocultured with MSCs.
FSCs can be isolated from cord blood or extraembryonic tissues have unique therapeutic advantages and disadvantages and have also been referred to as intermediaries between ESCs and ASCs [44]. Experimental studies show that transplanted human fetal neural stem cells have the ability to survive, differentiate, and migrate towards middle cerebral artery occlusion induced lesions in the ischemic rat cerebral cortex [45]. Similar studies have shown that human umbilical cord blood neural stem cells can also migrate to sites of damage and survive within the central nervous system (CNS) environment of the rat model [46]. When grafted into the developing brain, fetus-derived stem cells also have the ability to migrate to sites of injury and to differentiate into host cells specific to the target region [47]. The potential of fetal neural stem cell therapy should be weighed against limitations in cell expansion and the clinical hurdle of tumorigenicity [48, 49]. A brief synopsis of advantages and disadvantages of FSCs for the treatment of neurodegenerative disease is included in Table 2.
Table 2 Advantages and disadvantages of fetal stem cells for the treating neurodegenerative disease
| Stem Cell Type | Advantages | Disadvantages | Refs |
|---|---|---|---|
| Fetal Stem Cells from fetal tissues (e.g. fetal blood, fetal bone marrow, fetal liver, and fetal pancreas) |
- Favorable in-vivo tissue repair capacity, cell size, growth kinetics, and differentiation capacity - More primitive and possess greater multi-potentiality than adult stem cells - Better candidates for iPSCs reprogramming than somatic cells - Favorable pre-clinical outcomes for the treatment of osteogenesis imperfecta (OI) and Duchenne muscular dystrophy (DMD) |
- Ethical controversy |
13, 44, 64, 65, 148, 149 |
| Fetal Stem Cells from extraembryonic tissues (e.g. umbilical cord blood, Wharton’s Jelly, amniotic fluid, placenta) |
- Favorable pre-clinical outcomes for the treatment of Parkinson’s disease, malignant and non-malignant blood disorders, Diabetes Mellitus Type 2, and tissue injury - Reduced graft versus host disease - Anti-inflammatory and cell survival promoting secretions - Less restrictions in HLA compatibility - More primitive HSCs with greater proliferative potential than BM-HSCs - Advantages over ESCs and stem cells derived from adult bone marrow including unlimited source of UBC, lower risk of transmitting infections, immediate availability, greater tolerance of human leukocyte antigen HLA disparity and lower incidence of inducing severe graft-versus-host disease (GVHD) |
- Limited supply - Impossible to collect more donor cells in cases of engraftment failure or disease recurrence - High storage costs |
13, 44, 148, 150 |
The disadvantages associated with iPSCs, MSCs, FSCs, and ESCs have led to the consideration of alternatives such as ASCs. With regard to brain repair, unlike ESCs, ASCs offer the potential for transplantation without ethical dilemmas. Neurodegenerative disease may be treated with a variety of ASCs including NSCs, MSCs, HSCs, and stem cells from umbilical cord blood (UCB) [50-53]. Experimental evidence indicates that the mentioned stem cells can differentiate into neurons and glia [54-56]. However, there are distinct advantages of some ASC sources over others.
Adult neural stem cells are multipotent cells found within the subventricular zone (SVZ) of the olfactory bulb and the hippocampal dentate gyrus of the adult brain that can differentiate into neurons, glia, astroglia, and oligodendrocytes [57-61]. The advantages of adult neural stem cells are in the lack of ethical concerns associated with their use, their ability to be cultured, their potential for autologous transplantation, and their predetermined tendency to differentiate into cells of the neural lineage [62-65]. The disadvantages associated with adult neural stem cells are in the limited number of available cells, difficulty in expansion, cell senescence after several passages, paucity of clinical trial data, and their tendency to form tumors [62, 66]. These advantages and disadvantages should be considered within the framework of the relationships between adult NSCs and their niche, their cell compartment, and niche factors influencing their behavior [66, 67]. A brief synopsis of advantages and disadvantages of ASCs for the treatment of neurodegenerative diseases is included in Table 3.
Table 3
| Stem Cell | Advantages | Disadvantages | Refs |
|---|---|---|---|
| Adult HSCs |
- No ethical controversy |
- Limited Expansion - Limited Supply |
16, 52 |
| Adult NSCs |
- Animal studies indicate potential for improved cell engraftment and better motor function results than existing cellular therapies |
- Limited supply |
68, 73, 74, 101-107 |
| ADSC-non neural |
- Immune rejection unlikely - Abundant supply - Autologous transplant |
- Improvement in administration may be needed (e.g. to improve homing to damaged tissues or to minimize invasiveness) |
146 |
It is also prudent to keep in mind that a damaged neuron in need of repair or replacement is part of a functionally and morphologically complex network with tens of thousands of connections with other neurons; hence, replacing a neuron with a functional equivalent is insufficient unless this replacement is followed by integration into the appropriate functional circuit to recover previously compromised physical or mental activities [68]. In addition to cell replacement, stem cell therapies also offer potential benefits by mediating remyelination, exerting trophic actions, and modulating inflammation [69]. The therapeutic potential of adult NSCs for indications such as PD, ALS, AD, stroke, spinal cord injury, and other conditions needs to be studied in more detail with animal models and clinical trials [58, 69-71].
4 Blood Brain Barrier - Therapeutic Delivery
The skull, meninges membranes, cerebrospinal fluid, and the blood brain barrier (BBB) restrict access to the human brain. The BBB is a specialized non-permeable barrier with multiple components and unique efflux machinery that greatly complicates the treatment of brain tumors such as GBM. The BBB consists of endothelial cells united by tight junctions, astrocytic endfeet surrounding blood vessels, pericytes embedded in the vessel basement membranes, microglia, and neurons [72-74]. A graphic representation of the blood brain barrier is shown below (Figure 1):
The cellular junctions within the BBB are tight enough to enable strict selectivity in what crosses into the brain. For example, large hydrophilic molecules with a molecular mass of greater than 400-500 Da and more than 8-10 hydrogen bonds that are substrates for active efflux transporters at the BBB such as p-glycoprotein 1 cannot cross the BBB [74-77]. The vast majority of small molecule CNS drug candidates and essentially all large molecule CNS drug candidates including biotechnology products, recombinant proteins, monoclonal antibodies, RNA interference (RNAi) drugs, and gene therapy products do not cross the BBB [76]. Although numerous factors including molecular surface activity can influence BBB permeability to therapeutics, molecules that can penetrate the BBB generally do so through passive diffusion, carrier mediated transport, or receptor mediated transport [74, 78, 79]. BBB penetration by lipophilic Chlorpromazine Hydrochloride through adsorption to BBB endothelial cell capillaries serves as an example of transport by passive diffusion [79]. Movement of L-dihydroxy-phenylalanine (L-DOPA) through the BBB with the aid of the sodium-independent neutral amino acid transporter (LAT-1) is an example of carrier-mediated transport.
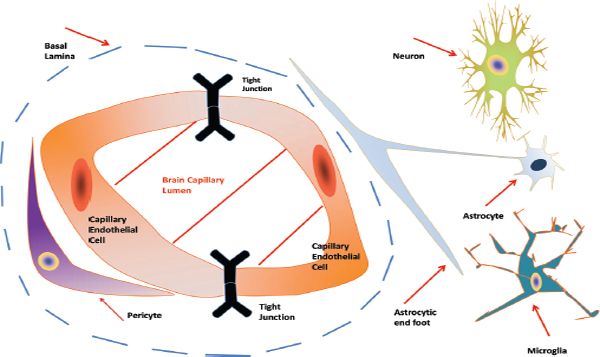
Figure 1 Representative diagram of the Blood Brain Barrier (BBB). Shown is a cross sectional view of the BBB. The components of the barrier consist of capillary endothelial cells united by tight junctions, astrocytic endfeet surrounding the blood vessels, pericytes embedded in the vessel basement membranes (BMs), microglia, and neurons.
Delivery of recombinant iduronate 2-sulfatase enzyme (IDS) through the BBB by IDS fusion to an IgG domain monoclonal antibody targeting the human insulin receptor is an example of receptor-mediated transport [80].
The brain’s impermeability to most therapeutics is problematic even for diseases that do not affect the brain. Metastatic tumors from tissues other than the brain can be found in the brain. Since most anticancer treatments cannot pass through the brain, the problem of treating secondary brain tumors originating from tissues other than the brain remains unresolved.
MSCs are immunosuppressive and can home to primary and metastatic tumors including breast, colon, ovarian, lung carcinomas, and gliomas [74, 81]. Some MSC therapies showed the potential to specifically target cancer stem cells (CSCs) and glioma cancer stem cells (gCSCs) that are critical to bulk tumor growth and recurrence [82, 83]. Human AT-MSCs have the ability to engraft into glioblastoma and target the vascular niche where gCSCs reside [84, 85]. Marrow-isolated adult multilineage inducible (MIAMI) are a subpopulation of mesenchymal stromal cells (MSCs) that have been used as therapeutic delivery vectors due to their ability to migrate to brain tumors and efficiently incorporate lipid nanocapsules (LNCs) without changing their stem cell properties or tumor-tropism [86]. Combination therapies including a recent study with mesenchymal stem cells secreting necrosis factor-related apoptosis-inducing ligand (TRAIL) and temozolomide (TMZ) demonstrated higher antitumor effects using an in vitro coculture system and an in vivo experimental xenografted mouse model than TMZ or MSCs secreting TRAIL alone suggesting an augmentation of caspase-mediated tumor apoptosis through death receptor 5 (DR5) upregulation and downregulation of antiapoptotic proteins [87].
NSCs have also been used as therapeutic delivery vectors for the treatment of brain tumors due to their patented ability to locally target diffuse metastatic brain tumors [88]. In particular, human fetal stem cells loaded with oncolytic viruses (OVs) have demonstrated improved survival in mouse animal models when compared with controls involving OVs without fetal stem cells [89]. Fetal stem cells loaded with mesoporous silica nanoparticles (MSNs) also remained viable longer than fetal stem cells without MSNs once implanted into the mouse [90]. One can argue that these trial results warrant further phase I studies. An overview of the mentioned studies is included in (Table 4). Significant challenges associated with the treatment of gliomas, and especially glioblastomas, make these cancers the basis to test whether stem cells can be effective in brain tumor treatment.
There are also several strategies by which glioma and glioblastoma therapeutics can be delivered past the BBB (Table 5). The BBB may be bypassed with intracerebral, intrathecal, intracranial, or intraventricular drug administration during surgery [74, 91]. One advantage of surgery would be very specific dosing by placing therapeutics near or within tumors [92-94]. Although surgical resection with chemotherapy is the standard treatment for high grade glioma, the disadvantage of invasive surgical delivery are surgical complications and the disruption of the BBB that can cause unwanted side effects linked to chronic neuropathologic changes [94, 95]. Other methods of therapeutic delivery through the BBB such as the loosening of endothelial cell tight junctions with ultrasound offer the advantage of less invasiveness than surgery by reversibly opening the BBB but carry the disadvantage of translational difficulty to fit the thickness and variability of the human skull and reduced control over dosage [96]. Chemical modifications to drugs including lipidation, glycosylation, carrier linkage, liposome loading, and other modifications present the advantage of improving drug permeability through the BBB but also have the drawback of creating the possibility of accumulation of drug metabolites within the brain that can lead to toxicity [97].
Table 4 Stem cells used in drug delivery for treating brain tumors
| Stem Cell | Sources | Tumor | Models | Therapeutic Modalities | Outcomes | Refs |
|---|---|---|---|---|---|---|
| MSC | Bone Marrow | Glioblastoma | CD IGS-Rat | Suicide gene cytosine deaminase::uracil phosphor-ribosyltransferase | Complete tumor regression | 151 |
| Adipose | Glioblastoma | CD IGS-Rat | Suicide gene cytosine deaminase:uracil phosphor-ribosyltransferase | Complete tumor regression | 151 | |
| MSC | Human Bone Marrow | Glioma | Male athymic mice | TMZ and MSCs secreting TRAIL | Augmentation of tumor apoptosis | 87 |
| MIAMI | Bone Marrow | Glioblastoma | Nude mouse | MIAMI cells with ferrociphenol | Cytotoxic effect on U87MG cells | 86 |
| NSC | Fetal brain cell line | Glioma | Athymic Nude Mouse | HB1.F3.CD cells loaded with CRAd-Survivin-pk7 | Improved mouse survival | 89 |
| NSC | Fetal brain Cell line | Glioma | Mouse | MSN-Dox-loaded HB1.F3.CD cells | Improved mouse survival and prolonged HB1.F3.CD cell viability | 90 |
Table 5 Therapeutic delivery for glioma treatment
| Methods | Therapies | References |
|---|---|---|
| Lipidation | Chemical | 74 |
| Glycosylation | Chemical | 74, 152 |
| Liposome loading | Chemical | 74, 153, 154 |
| Nanoparticle loading | Chemical | 74, 155 |
| Antibody Coupling | Chemical | 74, 156, 157 |
| BCNU Wafers | Chemical | 158 |
| Intracranial, | Chemical/ Biologics | 74, 159 |
| Intraventricular, | ||
| Intrahecal delivery | ||
| Intranodal delivery | Biologics/Vaccines | 160 |
| Ultrasound BBB disruption | Chemical/Biologics | 154, 161 |
| Intranasal delivery | Chemical/Biologics | 162 |
The table shows specific methods to deliver cells or drugs to the brain for the treatment of glioma and glioblastoma multiforme
Gliomas in the form of grade IV astrocytomas or GBM that originates as a primary tumor from the glial tissue within the brain is difficult to treat because it is separated from the rest of the human body by the BBB from the onset of tumor formation through tumor progression. When considering the limitations of surgery and chemotherapy for the treatment of highly aggressive tumors such as GBM, stem cells have the potential to offer benefits such as tumor targeting, tumor engraftment, participation in tumor stroma formation, ability to bind to radiotherapy resistant glioblastoma stem-like cells (GSCs), delivery of therapeutic ligands such as TNF-related apoptosis-inducing ligand (TRAIL), immunomodulation, and immunoenhancement.
Open-label trials have shown that transplants of fetal/embryonic midbrain tissue have exhibited some beneficial clinical effects while the alternative of neural grafting with fully differentiated adult neural cells has not become a standard treatment for several reasons including a limited supply of donor cells, unpredictable treatment benefits, and graft-related side effects [19]. Different kinds of embryonic stem cells possess unique advantages and disadvantages for the treatment of neural disorders.
In addition to providing basic NSC replacement through differentiation into NSCs, the aforementioned stem cell populations such as ADSCs and MSCs can also provide neurotrophic support leading to the mobilization of endogenous stem cells within the brain or to the therapeutic alteration of the brain microenvironment. Prior studies have also analysed ESC, HSC, and NSC properties at the transcriptional level to draw connections between “stemness” genes; these studies may lay the groundwork for the creation of genetically novel stem cell lines with unique therapeutic properties [98]. Novel genetic labelling methodologies can also shed light on native cell differentiation mechanisms [99].
5 Viral Gene Therapy
Treatment strategies for GBM using viruses have included the use of replication deficient viruses and replication competent oncolytic viruses (OV) [100, 101]. Replication deficient viruses can counteract GBM through the local activation of chemotherapeutic prodrugs, transfer of activated prodrugs through gap junctions via the “bystander effect”, and by activating the immune system [102]. Replication competent oncolytic viruses can counteract GBM by directly infecting tumor cells, replicating in tumor cells, and destroying tumor cells as well as by mediating the death of uninfected cells by blocking tumor blood supply and by activating virally encoded therapeutic transgenes [102, 103]. Although cancers have mechanisms for immunosuppression and immune detection evasion, vaccination offers the intriguing possibility of creating a sustained adaptive immune response against cancer as demonstrated by improvements in patient overall survival during the IMPACT trial that served as the basis for the approval of Sipuleucel-T by the FDA. Completed clinical trials data for the treatment of GBM with gene therapy and vaccination as adjuvants to surgical resection, chemotherapy, and radiotherapy are summarized in (Table 6).
The HSV-TK/GCV system has been widely used in phase I/II clinical trials to target rapidly dividing cancer cells. The Herpes Simplex Virus (HSV) is a cytotoxic oncolytic virus that can induce immune responses [104]. Thymidine kinase is a phosphotransferase enzyme responsible for the synthesis of DNA and cell division. GCV is a nucleoside analog that can be enzymatically phosphorylated by the herpes simplex virus thymidine kinase (HSV-TK) system to ganciclovir triphosphate. Suicide genes are enzymes that can convert prodrugs into therapeutics. Expression of the suicide gene encoding a viral enzyme, HSV-TK, results in the phosphorylation of the non-toxic prodrug, ganciclovir. The active, phosphorylated ganciclovir triphosphate is subsequently incorporated into the genome of rapidly dividing TK+ GBM tumor cells in vivo thereby leading to the selective elimination of malignant target cells [105, 106].
Table 6 Completed clinical trials for GBM
| Trial Phase | Disease | Design | Delivery and Dose | Results | PFS, mo. | OS, mo. | Year/Reference |
|---|---|---|---|---|---|---|---|
| I/II | Recurrent GBM | PA 317 cells delivering HSV-TK and IL-2 retroviral vector | 3×108-109 cells i.t. or into resection cavity and 5 mg/kg GCV i.v. twice a day for 14 days | No serious adverse events; tumor transduction as well as local and systemic type 1 T helper cell cytokine elevation | 4.5 | 7.5 | 2005 107 |
| II | EGFR vIII GBM | EGFR vIII peptide vaccine + TMZ STD or TMZ DI + XRT | 150 μg i.d. vaccine biweekly for 6 weeks after initial XRT | No serious adverse events; allergic reactions in TMZ DI cohort | 15.2 | 23.6 | 2011 163 |
| I | Recurrent and ND-GBM | Peptide vaccine associated with CSCs loaded onto autologous DCs | i.d. vaccine biweekly for 6 weeks after radiation or surgery in the axillary region | No serious adverse events; Grade 1 and 2 adverse events | 16.9 | 38.4 | 2013 164 |
| II | ND-GBM | Tumor lysates loaded onto DCs treated with TNF α and PGE2 | 1* 107 DC cells intranodal cervical lymph node injection every 2 weeks for 6 weeks after surgery + chemoradiotherapy | No serious adverse events; Grade 2 unilateral neck pain for 2 weeks in 1 patient after 1 cervical lymph node injection | 9.5 | 28 | 2011 165 |
| III | ND-GBM | PA 317 cells delivering HSV-TK and IL-2 retroviral vector | 1* 109 cells into resection cavity and 5 mg/kg GCV i.v. twice a day for 14 days and radiotherapy for 6 weeks | No improvement in progression free survival or median survival compared with surgical resection and radiotherapy control | 6.0 | 12.0 | 2000 113 |
| I/II | Recurrent GBM | PA317 cells delivering HSV-TK retroviral vector i.v. twice a day for 14 days | 1.5 × 108 -3 × 108 cells into resection cavity and 5 mg/kg GCV | No serious adverse events | N/A | 8.6 | 1999 166 |
| I/II | Recurrent GBM | NDV-HUJ | 0.1-11 billion infectious dose and 3 cycles of 55 billion infectious units i.v. or 2* 11 billion infectious dose units/weeki.v. | Maximum tolerated dose not reached; 1/11 CR, not durable; virus recoverable from body fluids | N/A (TTP = 6.5 mo) | 9.25 | 2006 112 |
| IB | Recurrent GBM | G207: ICP6-inactivated and ICP34.5-deleted HSV | 1.5 * 108 pfu i.t. 2-5 days before surgery + 1*109pfu into resection cavity | No dose-limiting toxicities; no CR or PR; immune cell infiltration post treatment | N/A (TTP = 3.0 mo) | 6.6 (OS = 23.0 mo from diagnosis) | 2009 167 |
GBM, glioblastoma multiforme; PFS, progression free survival; OS, overall survival; ND-GBM, newly diagnosed GBM; CR, complete response; PR, partial response; i.t., intrathecal; i.v., intravenous; EGFRvIII, epidermal growth factor receptor variant III tumor-specific mutation expressed in GBM; CSCs, cancer stem cells; DCs, dendritic cells; TNF α tumor necrosis factor alpha; PGE2, prostaglandin E2; TMZ, temozolomide; TMS STD, temozolomide standard dose; TMZ DI, temozolomide dose intensified; XRT, external beam radiotherapy; TTP, time to progression; PFU, plaque forming units.
A recent phase I/II trial used the HSV Type 1-Interleukin 2 (IL-2) gene therapy or HSV-TK/ IL-2 gene therapy and the drug Ganciclovir (GCV) to treat patients with recurring GBM [107]. This was the first clinical protocol for recurrent GBM combining the suicide gene (HSV-TK) delivery with a cytokine gene (IL-2). In this study, retroviral vector and vector producing cells (RVPCs) were derived from a single clone of the PA317 packaging cell line and expressed two therapeutic genes, human IL-2 and HSV-TK transcribed from the 5’ viral long terminal repeat and separated by an internal ribosome entry site (IRES) of encephalomyocarditis virus sequence and the antibiotic resistance gene neomycin phosphotransferase transcribed from an internal simian virus 40 (SV40) early promoter [107].
The outcomes of the this study among 12 patients showed that RPVC injection carrying the suicide gene HSV-TK and the cytokine gene IL-2 is safe up to 1*109 RVPCs, provides effective transduction of therapeutic genes to the target tumor cells, activates a cytokine response with a tumor response in 50% of the cases, and partial response in 16.7% of the cases including one patient with a disappearance of a distant noninjected tumor mass [107]. The safety of this treatment is notable considering that IL-2/HSV-TK delivery by intratumoral injection of xenographic RVPC elicited local inflammation in the CNS and that adverse events were mild and mainly associated with GCV administration [102]. Neurological adverse events were related to disease progression [102]. Unfortunately, two patients died from pulmonary embolisms at 2 and 11 months after first injection of gene therapy [102]. These patients had a marked increase of plasma cytokine levels after gene therapy, which is also associated with long-term tumor size reduction and clinical response in one of the patients [102]. While high-grade glioma patients are at increased risk for deep vein thrombosis (DVT) and pulmonary embolism (PE), the activation of the cytokine cascade after gene therapy which was associated with long-term tumor size reduction in one patient, may have also contributed to thromboembolic events [107-109].
In addition to the HSV-TK system utilizing retroviral PA317 cells, some of the other viral treatment modalities for gliomas and glioblastomas included the HSV-TK system with an adenoviral vector (Adv-HSV-TK), Newcastle Disease Virus (NDV), recombinant HSVs such as HSV1716 with deletions of the RL1 gene, and Herpes Simplex Virus G207 (HSV G 207) with deletions of the RL1 gene as well as ICP6 gene inactivation. Deletions of the RL1 gene were performed in order to make the recombinant HSVs such as HSV1716 non-neurovirulent [110]. Recombinant HSV G 207 featured deletions of the RL1 gene that prevented HSV from infecting non-dividing neural cells and also ICP6 gene inactivation as an extra safety measure to protect non-dividing healthy neural brain cells from unwanted HSV infection; the ICP6 gene encodes HSV ribonucleotide reductase, a key enzyme for viral DNA synthesis in non-dividing cells but not in dividing cells [111]. Another phase I/II trial for the treatment of recurrent GBM using the tumor selective HUJ (Hebrew University Jerusalem) strain of the oncolytic NDV attempted to assess the safety and tumor response of an accelerated dose escalation intrapatient intravenous infusion protocol with molecular imaging. Trial results among 11 patients with recurrent GBM indicated that i.v. dose escalation from 11-55 BIU (Billion Infectious Units) where (1 BIU= 1 *109 EID5050% egg infectious dose) of Hebrew University Jerusalem-New Castle Disease Virus (HUJ-NDV) was relatively well tolerated with five patients showing fever probably related to treatment [112]. Trial results also showed complete tumor response for one patient and featured overall survival ranging from 3 to 66 weeks [112].
In a Phase III clinical trial for untreated GBM, Rainov et al treated 248 patients with surgical resection and radiotherapy, or surgical resection with radiotherapy, HSV-TK, and GCV at the time of surgery. Gene therapy treatment was safe but no difference for overall safety and disease progression was observed between the control and the gene therapy group [113].
Another open label, randomized, parallel-group, multicenter phase III trial including 250 patients with supratentorial GBM amenable to resection compared gene therapy with sitimagene ceradenovec, a first generation replication-deficient adenovirus against the standard of care that included surgical resection, chemotherapy, and in some countries temozolomide administered at the discretion of the physician [114]. In the experimental group, 124 patients were given a one-time treatment consisting of between 30 to 70 perilesional injections of 100 micro liters of 1* 1012 viral particles of sitimagene ceradenovec containing cDNA for Herpes Simplex Virus-thymidine kinase with a blunt needle for up to 2 cm into the tumor wall following complete tumor resection [114]. The experimental group experienced 5 patient exclusions from intention to treat analysis, 5 days were given for transduction and then 5 mg/kg of ganciclovir were administered intravenously twice a day from 5 days to 19 days after the operation [114]. In the standard of care group, 126 patients were treated with complete tumor resection followed by 30 fractions to tumor volume of 60 Gy radiotherapy with a 2 cm margin or with complete tumor resection with radiochemotherapy per the Stupp protocol depending on temozolomide availability [114, 115]. The standard of care group experienced 9 patient exclusions from intention to treat analyses with 117 patients achieving the composite primary endpoint of time to death or re-intervention, where re-intervention was defined as any kind of treatment including surgery, radiotherapy, or chemotherapy given to extend survival when the tumor recurred or progressed [114]. Analysis of the composite primary endpoint was performed with a covariate Cox proportional hazards model with terms for treatment, age, baseline Karnofsky score, extent of tumor resection, MGMT methylation status as a fixed covariate, and temozolomide use as a time-dependent covariate because each of these covariates had an important effect on trial outcomes [114]. Ahazard ratio was calculated where a hazard ratio greater than one indicated a benefit for sitimagene ceradenovec. Statistical analysis showed that sitimagene ceradenovec improved median time to death or re-intervention regardless of temozolomide use (236 patients; HR 1*53, 95% CI 1*13-2* 07; p= 0* 0055). Sitimagene ceradenovec also demonstrated a statistically significant improvement on median time to time or re-intervention in the subgroup of patients who did not use temozolomide (n=102 patients, HR 1*58, 1*02-2*45; p=0 * 0398) [114]. Sitimagene ceradenovec had an even greater effect on the primary endpoint in non-methylated patients than it did in the whole cohort (HR 1*72, 95% CI 1*15-2*56, p=0*008) 114. Overall, the median time to death or re-intervention was longer in the group receiving sitimagene ceradenovec treatment compared to the standard of care group (268 days, 210-313; hazard ratio [HR] 1*53, 95% CI 1*13-2*07; p=0*006). However, there was no statistically significant difference (p=0*31) for the overall survival between the two groups (median 497 days, 95% CI369-574 for the experimental group vs 452 days, 95% CI 437-558 for the control group; HR 1*18, 95% CI 0*86-1*61) [114]. Although the most common adverse effects of sitimagene ceradenovec were hemiparesis, hyponatraemia, and seizures, the overall risk benefit ratio of the experimental treatment appears to be positive and indicates an advancement for the field of surgically applied and virally mediated local gene therapy [114]. However, the discrepancy between significant improvement in time to death or progression without a significant improvement in overall survival may indicate poor transduction or poor delivery, thereby providing impetus for the development of alternative delivery and transduction mechanisms.
6 Cancer Stem Cells (CSCs)
There is increasing evidence showing a small subpopulation of cells within solid and soluble malignant tumors known as CSCs that are responsible for the initiation and growth of malignancy [82, 83]. Recent studies have identified a subpopulation of brain tumor stem cells in adult GBMs known as CSCs that can self-renew, express genes associated with NSCs, generate daughter cells of different phenotypes from one mother cell, and differentiate into a phenotypically diverse population of cells present in the initial GBM [82, 83]. There is also evidence that CSCs do not necessarily need to arise from stem cells but can also come from lineage-restricted progenitor cells, for example, expression of the ras and myc oncogenes in lineage-restricted Oligodendrocyte-type-2 astrocyte (O-2A) progenitor cells yields cells that readily form tumors when transplanted in vivo [82, 116].
7 Targeting Cancer Stem Cells
An understanding of CSCs and their relationship to the bulk tumor is important because cancer stem cells play a key role in tumor recurrence [83]. Cancer has the ability to evade or inactivate the immune response through a number of mechanisms including mutations in genes encoding target antigens, loss of antigen expression, or immunosuppressive actions such as the secretion of transforming growth factor-β (TGFβ), vascular endothelial growth factor (VEGF), galectin, or indolamine 2,3-dioxygenase (IDO) and/or through the recruitment of immunosuppressive immune cells [117, 119]. Clinical trials data for adjuvant cancer vaccines designed to illicit an immune response to tumor antigens, tumor-associated antigens, and prevent further growth of cancer refractory to conventional therapies such as surgeries, radiation therapy, and chemotherapy is summarized in (Table 6) [118]. The first generation of patient-isolated or ex-vivo generated monocyte-derived dendritic cell cancer vaccines was loaded with tumor lysates, recombinant tumor antigens, or synthetic peptides and proven to be safe in early clinical trials but also encountered some limitations such as limited tumor regression rate, lack of linear dose-response effects, and inability to fully accommodate immune target complexity [118]. To circumvent the mentioned and to create dendritic cells with maximal immunogenicity, currently produced dendritic cell vaccines are matured and activated in the presence of specific cytokines, pathogen-derived agonists, toll-like receptor ligands and exposed to modifications of specific genes such as Egr2 gene silencing [118]. Another important consideration for future studies may be the altered expression of several MicroRNAs (miRNAs) identified in gliomas. MiRNAs are endogenously expressed small noncoding RNAs that regulate genes, post-transcriptionally; it is possible that more insights on the relationship between miRNAs and the tumor microenvironment can lead to the development of better cancer treatments.
8 Stem Cells and Anti-Cancer Vaccines
Dendritic cells (DCs) are antigen presenting cells (APCs) found in the mammalian immune system assembled within the bone marrow in response to fms-like tyrosine kinase-3 ligand (Ftl3L) and granulocyte-macrophage colonystimulating factor (GM-CSF) [117]. DC precursors and immature DCs found in the human blood are lineage negative (CD3~, CD14”, CD19”, CD56”) HLA-DR+mononuclear cells that can be categorized into plasmacytoid dendritic cells (PDCs) CD11c~, CD123hi and myeloid dendritic cells (MDCs) CD11c+, CD123’° [117]. PDCs are the principal interferon alpha producing cells in the human body and can activate antitumor and antiviral responses, however, their immunotherapeutic potential is largely unknown because they are difficult to obtain in large quantities [120, 121].
DC-based cancer vaccines focus on active vaccination against tumorspecific antigens [117]. As of 2014, 289 clinical studies of DC-based cancer vaccines are registered and under investigation (2014, http://www.clinicaltrials.gov). Among the 289 cases, 2 are in phase IV, 6 in phase III, 3 in phases II and III, 74 in phase II, 76 in phases I and II, 109 in phase I, and 3 in phase 0, underscoring the potential clinical significance of this therapy [118].
9 Combination Therapies
Active immunotherapies against cancer work by harnessing the cytotoxic T lymphocyte (CTL) response by several means that may overlap: changing the tumor microenvironment to stimulate immune cell function, creating large amounts of tumor-specific T cells for adoptive cell transfer (ACT), administering immune stimulating cytokines or immune modulating antibodies to induce nonspecific immune responses against tumors, and administering new cancer vaccines to induce immunity against specific tumor antigens.
10 Chimeric Antigen Receptor
Chimeric antigen receptor (CAR) T-cells were developed in an effort to give T-cells antigen specificity by fusing an antibody-derived targeting domain with T-cell signalling domains. In a seminal study, Eshhar et al fused a single chain of an Fv (scFv) antibody molecule to the γ chain of the Fc receptor or to the ζ of the CD3 complex [122]. These first generation CAR T-cells displayed several immunotherapeutic advantages including an MHC independent targetbinding mechanism, target-binding affinity much higher than T-cell receptors (TCRs), antibody type specificity, and IL-2 signalling leading to target cell lysis [122]. CAR T-cells may become an immunotherapy treatment option for patients with refractory brain tumors due to their ability to enter the brain, and target glioblastoma tumor receptors such as EGFR [123].
Second and third generation CAR T-cells have been engineered to improve potency and targeting. Further understanding of co-stimulatory signals including CD80, CD86, CD28, 4-1BB, and OX40 has lead to the development of CAR T-cells with more target cytotoxicity and less T-cell anergy or T-cell apoptosis than their first generation counterparts [124-127].
HLA-independent recognition of target antigens and the delivery of a large population of tumor antigen-specific T-cells are some of the advantages of CAR T-cell therapy. On the other hand, there are also important disadvantages including “off-tumor/on-target toxicity” and cytokine-release syndrome.
Cytokine-release syndrome is driven by inflammation inducing cytokines including IFNγ, TNFβ, IL-2, and IL-6 [128-131]. Studies with an anti IL-6 monoclonal antibody, toclizumab, in glucocorticoid resistant GvHD showed the biological effects of IL-6 [132]. T-cell expansion and cytokine-release syndrome seem to be related as patients present with fever, myalgias, nausea, anorexia, and various complications [129].
In a recent clinical trial, CAR T-cells with specificity for CD19 injected into two patients with chronic lymphocytic leukemia (CLL) lead to complete remission in both patients, T- cell expansion 1000 times higher than the initial treatment level, and the presence of CAR T-cells in the cerebrospinal fluid [131]. Unfortunately, this trial also reported grade 3 or 4 adverse events, cytokine-release syndrome, and B-cell aplasia [131].
There are medical limitations that limit the use of CAR T-cell therapy. CAR T-cell therapy cannot be effective without the immunogenicity of the target cells and CSCs are attractive future target cell candidates. Although most data on CAR T-cell therapy is limited to haematological malignancies; CAR T-cell therapy does have the potential to translate to the treatment of solid brain tumors such as neuroblastoma [133]. The mechanisms behind CAR T-cell therapy adverse events must be addressed and this may lead to engineering of other cells including iNKT and NK cells.
11 Conclusions
The difficulty in treating GBM seems to be attributable to tumor location, tumor heterogeneity, and the actions of gCSCs in generating and regenerating tumor growth [134]. The BBB, which isolates GBM tumors from the rest of the body, complicates the delivery of therapeutics and reduces immune system access. Stem cells may be able to significantly improve therapeutic delivery due to their tumor-tropism and ability to cross the BBB.
GBM is able to evade the immune response. Research on BBB permeability may improve therapeutic targeting, timing, and transport. Various kinds of stem cells have been studied as potential vehicles for the delivery of suicide genes and drugs to the tumor bed. Although MSCs can be easily expanded and harvested, disadvantages associated with possible transformation need to be addressed [135].
While gene therapy has been extensively studied for GBM, phase III trials have failed to demonstrate significant advantages. This may be attributable to poor drug delivery and inconsistent transduction [136]. Therapeutic modalities enhancing patient immune response such as vaccines targeting gCSCs and CAR T-cells may have great potential. CAR T-cells are briefly discussed due to comparative ease of delivery to the brain [123]. Favorable results for cytoxocity and increased survival in animal models from in vivo and in vitro studies present MSCs and NSCs as potential delivery vehicles for the treatment of GBM.
GBM needs to be approached from different angles given its heterogeneity, localization, and resistance to treatment. Stem cell research can be an important starting point given the limitations of current treatment.
References
[1] Wen, P. Y., and S. Kesari. 2008. Malignant gliomas in adults. N Engl. J. Med. 359: 492-507.
[2] Lee, J. K., K. M. Joo, J. Lee, Y. Yoon, D. H. Nam. 2014. Targeting the epithelial to mesenchymal transition in glioblastoma: the emerging role of MET signaling. Onco Targets Ther. 7: 1933-1944.
[3] Wilson, T. A., M. A. Karajannis, D. H. Harter. 2014. Glioblastoma multiforme: State of the art and future therapeutics. Surg. Neurol. Int. 5: 64.
[4] Ferlay, J., H. R. Shin, F. Bray, D. Forman, C. Mathers, D. M. Parkin. 2010. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer. 127: 2893-2917.
[5] Serão, N. V. L., K. R. Delfino, B. R. Southey, J. E. Beever, and S. L. Rodriguez-Zas. 2011. Cell cycle and aging, morphogenesis, and response to stimuli genes are individualized biomarkers of glioblastoma progression and survival. BMC Med. Genomics 4: 1-21.
[6] Dai, C., and E. Holland. 2005. Astrocyte differentiation states and glioma formation. In Glioblastoma Multiforme. J. Markert, V. T. DeVita, S. A. Rosenberg, and S. Hellman, eds. Jones and Barlett Publishers, Subury, MA. p. 1-316.
[7] Kleihues, P., and H. Ohgaki. Primary and secondary glioblastomas: from concept to clinical diagnosis. Neuro-Oncology 1998: 44-51.
[8] Morizane, A., J. Y. Li, P. Brundin. 2008. From bench to bedside: the potential of stem cells for the treatment of Parkinson’s disease. Cell Tissue Res. 331: 323-336.
[9] Mouhieddine, T. H., F. H. Kobeissy, M. Itani, A. Nokkari, and K. K. W. Wang. 2014. Stem cells in neuroinjury and neurodegenerative disorders: challenges and future neurotherapeutic prospects. Neural Regen. Res. 9: 901-906.
[10] Feng, Z., and F. Gao. 2012. Stem cell challenges in the treatment of neurodegenerative disease. CNS Neurosci. Ther. 18: 142-148.
[11] Kim, S. U., H. J. Lee, and Y. B. Kim. 2013. Neural stem cell-based treatment for neurodegenerative diseases. Neuropathology 33: 491-504.
[12] Wang, S., H. Cheng, G. Dai, X. Wang, R. Hua, X. Liu, P. Wang, G. Chen, W. Yue, Y. An. 2013. Umbilical cord mesenchymal stem cell transplantation significantly improves neurological function in patients with sequelae of traumatic brain injury. Brain Res. 1532: 76-84.
[13] Achyut, B. R., N. R. S. Varma, A. S. Arbab. 2014. Application of umbilical cord blood derived stem cells in diseases of the nervous system. J. Stem Cell Res. Ther. 4: 202.
[14] Ali, H., N. Bayatti, S. Lindsay, A. A. Dashti, F. Al-Mulla. 2013. Directed differentiation of umbilical cord blood stem cells into cortical GABAergic neurons. Acta Neurobiol. Exp. 73: 250-259.
[15] Volarevic, V., N. Arsenijevic, M. L. Lukic, and M. Stojkovic. 2011. Concise review: mesenchymal stem cell treatment of the complications of diabetes mellitus. Stem Cells 29: 5-10.
[16] Sigurjonsson, O. E., M. C. Perreault, T. Egeland, J. C. Glover. 2005. Adult human hematopoietic stem cells produce neurons efficiently in the regenerating chicken embryo spinal cord. Proc. Natl. Acad. Sci. USA 102: 5227-5232.
[17] Pang, Z. P., N. Yang, T. Vierbuchen, A. Ostermeier, D. R. Fuentes, T. Q. Yang, A. Citri, V. Sebastiano, S. Marro, T. C. Sudhof, M. Wernig. 2011. Induction of human neuronal cells by defined transcription factors. Nature 476: 220-223.
[18] Liu, X., F. Li, E. A. Stubblefield, B. Blanchard, T. L. Richards, G. A. Larson, Y. He, Q. Huang, A. C. Tan, D. Zhang, T. A. Benke, J. R. Sladek, N. R. Zahniser, C. Y. Li. 2012. Direct reprogramming of human fibroblasts into dopaminergic neuron-like cells. Cell Res. 22: 321-32.
[19] Takahashi, K., and S. Yamanaka. 2006. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126: 663-676.
[20] Sivapatham, R., and X. Zeng. 2014. Generation and characterization of patient-specific induced pluripotent stem cell for disease modeling. Methods Mol. Biol. 2014: 1-20.
[21] Streckfuss-Bomeke, K., F. Wolf, A. Azizian, M. Stauske, M. Tiburcy, S. Wagner, D. Hubscher, R. Dressel, S. Chen, J. Jende, G. Wulf, V. Lorenz, M. P. Schon, L. S. Maier, W. H. Zimmermann, G. Hasenfuss, and K. Guan. 2013. Comparative study of human-induced pluripotent stem cells derived from bone marrow cells, hair keratinocytes, and skin fibroblasts. Eur. Heart J. 34: 2618-2629.
[22] Sunberg, M., H. Bogetofte, T. Lawson, J. Jansson, G. Smith, A. Astradsson, M. Moore, T. Osborn, O. Cooper, R. Spealman, P. Hallet, O. Isacson. 2013. Improved cell therapy protocols for Parkinson’s disease based on differentiation efficiency and safety of hESC-, hiPSC-, and non-human primate iPSC-derived dopaminergic neurons. Stem Cells 31 :1548-1562.
[23] Buttery, P. C., and R. A. Barker. 2014. Treating Parkinson’s disease in the 21st century: Can stem cell transplantation compete? J. Comparative Neurol. 522: 2802-2816.
[24] Kim, D., C. H. Kim, J. I. Moon, Y. G. Chung, M. Y. Chang, B. S. Han, S. Ko, E. Yang, K. Y. Cha, R. Lanza, and K. S. Kim. 2009. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell 4: 472-476.
[25] Hou, L. L., and T. Hong. 2012. Stem cells and neurodegenerative diseases. CNS Neurosci. Ther. 51:287-294.
[26] Soldner, F., D. Hockemeyer, C. Beard, Q. Gao, G.W. Bell, E.G. Cook, G. Hargus, A. Blak, O. Cooper, M. Mitalipova, O. Isacson, and R. Jaenisch. 2009. Parkinson’s disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell 136: 964-977.
[27] Huang, G. T.-J., S. Gronthos, and S. Shi. 2009. Mesenchymal stem cells derived from dental tissues vs. those from other sources. J. Dent. Res. 88: 792-806.
[28] Caplan, A. I., and J. E. Dennis. 2006. Mesenchymal stem cells as trophic mediators. J. Cell Biochem. 98: 1076-1084.
[29] Phiney, D. G., and D. J. Prockop. 2007. Mesenchymal stem/multipotent stromal cells: the state of transdifferentiation and modes of tissue repaircurrent views. Stem Cells 25: 2896-2902.
[30] Pittenger, M. F., A. M. Mackay, S. C. Beck, R. K. Jaiswal, R. Douglas, J. D. Mosca, M. A. Moorman, D. W. Moorman, D. W. Simonetti, S. Craig, and D. R. Marshak.1999. Multilineage potential of adult human mesenchymal stem cells. Science 284: 143-147.
[31] Asakura, A., M. Komaki, M. A. Rudnicki. 2001. Muscle satellite cells are multipotential stem cells that exhibit myogenic, osteogenic, and adipogenic differentiation. Differentiation 68: 245-253.
[32] De Bari, C., F. Dell’Accio, P. Tylzanowski, F. P. Luyten. 2001. Multipotent mesenchymal stem cells from adult human synovial membrane. Arthritis Rheum. 44: 1928-1942.
[33] Akimoto, K., K. Kimura, M. Nagano, S. Takano, G.T. Salazar, T. Yamashita, and O. Ohneda. 2013. Umbilical cord blood-derived mesenchymal stem cells inhibit, but adipose tissue-derived mesenchymal stem cells promote, glioblastoma multiforme proliferation. Stem Cells Dev 22: 1370-1386.
[34] Nagano, M., K. Kimura, T. Yamashita, K. Ohneda, D. Nozawa, H. Hamada, H. Yoshikawa, N. Ochiai, and O. Ohneda. 2010. Hypoxia responsive mesenchymal stem cells derived from human umbilical cord blood are effective for bone repair. Stem Cells Dev 19: 1195-1210.
[35] Bieback, K., S. Kern, H. Klüter, and H. Eichler. 2004. Critical parameters for the isolation of mesenchymal stem cells from umbilical cord blood. Stem Cells 22: 625-634.
[36] Parolini, O., F. Alviano, G. P. Bagnara, G. Bilic, H. J. Buhring, M. Evangelista, S. Hennerbichler, B. Liu, M. Magatti, N. Mao, T. Miki, F. Marongiu, H. Nakajima, T. Nikaido, C. B. Portmann-Lanz, V. Sankar, M. Soncini, G. Stadler, D. Surbek, T. A. Takahashi, H. Redl, N. Sakuragawa, S. Wolbank, S. Zeisberger, A. Zisch, and S. C. Strom. 2008. Concise review: isolation and characterization of cells from human term placenta: outcome of the first international Workshop on Placenta Derived Stem Cells. Stem Cells 26: 300-311.
[37] Tran, T. C., K. Kimura, M. Nagano, T. Yamashita, K. Ohneda, H. Sugimori, F. Sato, Y. Sakakibara, H. Hamada, H. Yoshikawa. S. N. Hoang, and O. Ohneda. 2011. Identification of human placenta-derived mesenchymal stem cells involved in re-endothelialization. J. Cell. Physiol. 226: 224-235.
[38] De Coppi, P., G. Bartsch, M. M. Siddiqui, T. Xu, C. C. Santos, L. Perin, G. Mustoslavsky, A. C. Serre, E. Y. Snyder, J. J. Yoo, M. E. Furth, S. Soker, and A. Atala. 2007. Isolation of amniotic stem cell lines with potential for therapy. Nat. Biotechnol. 25: 100-106.
[39] Ciavarella, S., F. Dammacco, M. De Matteo, G. Loverro, and F. Silvestris. 2009. Umbilical cord mesenchymal stem cells: role of regulatory genes in their differentiation to osteoblasts. Stem Cells Dev. 18: 1211-1220.
[40] Castillo, M., K. Liu, L. Bonilla, and P. Rameshwar. 2007. The immune properties of mesenchymal stem cells. Int. J. Biomed. Sci. 3: 76-80.
[41] Mariotti, V., S. J. Greco, R. D. Mohan, G. R. Nahas, and P. Rameshwar. 2014. Stem cell in alternative treatment for brain tumors: potential for gene delivery. Mol. and Cell. Ther. 2: 1-10.
[42] Pendleton, C., Q. Li, D. A. Chesler, K. Yuan, H. Guerrero-Cazares, and A. Quinones-Hinojosa. 2013. Mesenchymal stem cells derived from adipose tissue vs bone marrow: in vitro comparison of their tropism towards gliomas. PLos One 8:e58198.
[43] Lamfers, M., S. Idema, F. van Milligen, T. Schouten, P. van der Valk, P. Vandertop, C. Dirven, and D. Noske. 2009. Homing properties of adipose-derived stem cells to intracerebral glioma and the effects of adenovirus infection. Cancer Lett. 274: 78-87.
[44] Abdulrazzak, H., D. Moschidou, G. Jones, and P.V. Guillot. 2010. Biological characteristics of stem cells from foetal, cord blood and extraembryonic tissues. J. R. Soc. Interface 7:S689-S706.
[45] Kelly, S., T. M. Bliss, A. K. Shah, G. H. Sun, M. Ma, W. C. Foo, J. Masel, M. A. Yenari, I. L. Weissman, N. Uchida, T. Palmer, and G. K. Steinberg. 2004. Transplanted human fetal neural stem cells survive, migrate, and differentiate in ischemic rat cerebral cortex. Proc. Natl. Acad. Sci. USA 101: 11839-11844.
[46] Jablonska, A., H. Kozlowska, I. Markiewicz, K. Domanska-Janik, and B. Lukomska. 2010. Transplantation of neural stem cells derived from human cord blood to the brain of adult and neonatal rats. Acta Neurobiol. Exp. 70: 337-350.
[47] Armstrong, R. J., C. Watts, C. N. Svendsen, S. B. Dunnett, and A. E. Rosser. 2000. Survival, neuronal differentiation, and fiber outgrowth of propagated human neural precursor grafts in an animal model of Huntington’s disease. Cell Transplant. 9: 55-64.
[48] Lee, A. S., C. Tang, M. S. Rao, I. L. Weissman, and J. C. Wu. 2013. Tumorigenicity as a clinical hurdle for pluripotent stem cell therapies. Nat. Med. 19: 998-1004.
[49] Chen, K. G., B. S. Mallon, R. D. G. McKay, and P. G. Robey. 2014. Human pluripotent stem cell culture: considerations for maintenance, expansion, and therapeutics. Cell Stem Cell 14: 13-26.
[50] Petit, G. H., T. T. Olsson, and P. Brundin. 2014. The future of cell therapies and brain repair: Parkinson’s disease. Neuropathol. Appl. Neurobiol. 40: 60-70.
[51] Park, D. Y., R. E. Mayle, R. L. Smith, I. Corcoran-Schwartz, A. I. Kharizi, and I. Cheng. 2013. Combined transplantation of human neuronal and mesenchymal stem cells following spinal cord injury. Global Spine J. 3: 1-6.
[52] Borlongan, C. V., L. E. Glover, N. Tajiri, Y. Kaneko, and T. B. Freeman. 2011. The great migration of bone marrow-derived stem cells toward the ischemic brain: therapeutic implications for stroke and other neurological disorders. Prog. Neurobiol. 95: 213-228. [53] Zhao, F., Y. Qu, H. Liu, B. Du, and D. Mu. 2014. Umbilical cord blood mesenchymal stem cells co-modified by TERT and BDNF: a neuroprotective therapy for neonatal hypoxic-ischemic brain damage. Int. J. Dev. Neurosci. 38: 147-154.
[54] Fu, L., L. Zhu, Y. Huang, T. D. Lee, S. J. Forman, C. C. Shih. 2008. Derivation of neural stem cells from mesenchymal stemcells: evidence for a bipotential stem cell population. Stem Cells Dev. 17: 1109-1121.
[55] Castano, J., P. Menendez, C. Bruzos-Cidon, M. Straccia, A. Sousa, L. Zabaleta, N. Vazquez, A. Zubiarrain, K. C. Sonntag, L. Ugedo, X. Carvajal-Vergara, J. M. Canals, M. Torrecilla, R. Sanchez-Pernaute, and A. Giorgetti. 2014. Fast and efficient neural conversion of human hematopoietic cells. Stem Cell Rep. 3: 1118-1131.
[56] Sun, T., and Q. H. Ma. 2013. Repairing neural injuries using human umbilical cord blood. Mol. Neurobiol. 47: 938-945.
[57] Sanai, N., A. Alvarez-Buyla, and M. S. Berger. 2005. Neural stem cells and the origin ofgliomas. N Engl. J. Med. 335: 811-822.
[58] Gage, F. H. 2000. Mammalian neural stem cells. Science 287: 1433-1438.
[59] Doetsch, F., I. Caille, D. A. Lim, J.M. Garcia-Verdugo, and A. Alvarez-Buylla. 1999. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell 97: 703-716.
[60] Sohur, U. S., J. G. Emsley, B. D. Mitchell, and J. D. Macklis. 2006. Adult neurogenesis and cellular brain repair with neural progenitors, precursors, and stem cells. Philos. Trans. R. Soc. Lond. B Biol. Sci. 361: 1477-1497.
[61] Emsley, J. G., B. D. Mitchell, G. Kempermann, and J. D. Macklis. 2005. Adult neurogenesis and repair of the adult CNS with neural progenitors, precursors, and stem cells. Prog. Neurobiol. 75: 321-341.
[62] Galvin, K. A. and D. G. Jones. 2006. Adult human neural stem cells for autologous cell replacement therapies for neurodegenerative disorders. NeuroRehabilitation 21: 255-265.
[63] Arsenijevic, Y., J. G. Villemure, J. F. Brunet, J. J. Bloch, N. Deglon, C. Kostic, A. Zurn, and P. Aebischer. 2001. Isolation of multipotent neural precursors residing in the cortex of the adult human brain. Exp. Neurol. 170: 48-62.
[64] Kukekov, V.G., E. D. Laywell, O. Suslov, K. Davies, B. Scheffler, L. B. Thomas, T. F. O’Brien, M. Kusakabe, and D. A. Steindler. Multipotent stem/progenitor cells with similar properties arise from two neurogenic regions of adult human brain. Exp. Neurol. 156: 333-344.
[65] Juengst, E., and M. Fossel. 2000. The ethics of embryonic stem cells— now and forever, cells without end. J. Am. Med. A 284: 3180-3184.
[66] Kornblum, H. I. 2007. Introduction to neural stem cells. Stroke 38: 810-816.
[67] Fuentealba, L. C., K. Obernier, and A. Alvarez-Buylla. 2012. Adult neural stem cells bridge their niche. Cell Stem Cell 10: 698-708.
[68] Chiu, A. Y., and M. S. Rao. 2011. Cell-based therapy for neural disorders-anticipating challenges. Neurotherapeutics 8 (4): 744-752.
[69] Lindvall, O., and Z. Kokaia. 2010. Stem cells in human neurodegenerative disorders-time for clinical translation? J. Clin. Invest. 120 (1): 29-40.
[70] De Filippis, L., and E. Binda. Concise review: self-renewal in the central nervous system: neural stem cells from embryo to adult. Stem Cells Transl. Med. 1 (4): 298-308.
[71] Dantuma, E., S. Merchant, K. Sugaya. 2010. Stem cells in human neurodegenerative disorders-time for clinical translation? J Clin. Invest. 1 (5): 37.
[72] Dubois, LG., L. Campanati, C. Righy, I. D’Andrea-Meira, T. C. Leite de Sampaio e Spohr, I. Porto-Carreiro, C. M. Pereira, J. Balca-Silva, S. A. Kahn, M. F. DosSantos, M. De Almeida Rabello Oliveira, A. Ximenesda Silva, M. C. Lopes, E. Faveret, E. Leandro, and G. V. Moura-Neto. 2014. Gliomas and the vascular fragility of the blood brain barrier. Front. Cell Neurosci. 8: 418.
[73] Lossinsky, A. S., and R.R. Schivers. 2004. Structural pathways for macromolecular and cellular transport across the blood-brain barrier during inflammatory conditions. Review. Histol. Histopathol. 19 (2): 535-564.
[74] Aleynik, A., K. M. Gernavage, Y. S. H. Mourad, L. S. Sherman, K. Liu, Y. A. Gubenko, and P. Rameshwar. 2014. Stem cell delivery of therapies for brain disorders. Clin. Transl. Med. 3: 24 doi:10.1186/2001-1326-3-24.
[75] Pardridge, W. M. 2006. Molecular Trojan horses for blood-brain barrier drug delivery. Discov. Med. 6 (5): 494-500.
[76] Pardridge, W., M. 2001. Brain Drug targeting and gene technologies. Jpn. J. Pharmacol. 87 (2): 97-103.
[77] Pardridge W. M. 2000. Blood-brain barrier drug targeting enables neuroprotection in brain ischemia following delayed intravenous administration of neurotrophins. In: Madame Curie Bioscience Database [Internet]. Landes Bioscience 2000, Austin, TX. Available from: http://www.ncbi.nlm.nih.gov/books/NBK5974/
[78] Aiken, R. 2014. Molecular neuro-oncology and the challenge of the blood-brain barrier. Semin. Oncol. 41 (4): 438-445.
[79] Seelig, A., R. Gottschlich, and R. M. Devant. 1994. A method to determine the ability of drugs to diffuse through the blood-brain barrier. PNAS 91: 68-72.
[80] Boado, R. J., E. Ka-Wai Hui, J. Zhiqiang Lu, and W. M. Pardridge. 2014. Insulin receptor antibody-iduronate 2-sulfatase fusion protein: pharmacokinetics, anti-drug antibody, and safety pharmacology in rhesus monkeys. Biotechnol. Bioeng. 111 (11): 2317-25.
[81] Reagan, M. R., and D. L. Kaplan. 2011. Concise review: mesenchymal stem cell tumor-homing: detection methods in disease model systems. Stem Cells 29 (6): 920-927.
[82] Jordan, C. T., M. L. Guzman, and M. Noble. 2006. Cancer stem cells. NEng. J. Med. 335: 1253-1261.
[83] Xiangpen, Y., J. Curtis, X. Yizhi, L. Gentao, S. Waschsmann-Hogiu, D. L. Farkas, K. L. Black, and J. S. Yu. 2004. Isolation of cancer stem cells from adult glioblastoma multiforme. Oncogene 23 (58): 9392-9400.
[84] Alieva, M., J. R. Bago, E. Aguilar, C. Soler-Botija, O. F. Vila, J. Molet, S. S. Gambhir, N. Rubio, and J. Blanco. 2012. Glioblastoma therapy with cytotoxic mesenchymal stromal cells optimized by bioluminescence imaging of tumor and therapeutic cell response. PloS One 7 (4):e35148.
[85] Altanerova, V., M. Cihova, M. Babic, B. Rychly, K. Ondicova, B. Mravec, and C. Altaner. 2012. Human adipose tissue-derived mesenchymal stem cells expressing yeast cytosinedeaminase::uracil phosphoribosyltransferase inhibit intracerebral rat glioblastoma. Int. J. Cancer 130 (10): 2455-2463.
[86] Roger, M., A. Clavreul, N. Trinh Huynh, C. Passirani, P. Schiller, A. Vessières, C. Montero-Meneia, and P. Menei. 2011. Ferrociphenol lipid nanocapsule delivery by mesenchymal stromal cells in brain tumor therapy. Int. J. Pharm. 423 (1): 63-68.
[87] Kim, S. M., J. S. Woo, C. H. Jeong, C. H. Ryu, J. D. Jang, and S. S. Jeun. 2014. Potential application of temozolomide in mesenchymal stem cell-based trail gene therapy against malignant glioma. Stem Cells Transl. Med. 3 (2): 172-182.
[88] Snyder, E. Y., X. O. Breakefield, K. S. Aboody, U. Herrlinger, and W. P. Lynch, inventor The Children’s MedicalCenter Corporation Boston, MA, USA; The General Hospital Corporation, Charlestown, MA, USA; Northeastern Ohio Universities College of Medicine, Rootstown, OH, USA___assignee. US Patent # 7186409. Neural stem cells and use thereof for brain tumor therapy. United States 2007.
[89] Ahmed, A. U., B. Thaci, A. L. Tobias, B. Auffinger, L. Zhang, Y. Cheng, C. K. Kim, C. Yunis, Y. Han, N. G. Alexiades, X. Fan, K. S. Aboody, and M. S. Lesniak. 2012. A preclinical evaluation of neural stem cell-based cell carrier for targeted antiglioma oncolytic virotherapy. JNCI 105 (13): 968-977.
[90] Cheng, Y., R. Morshed, S. H. Cheng, A. Tobias, B. Auffinger, D. A. Wainwright, L. Zhang, C. Yunis, Y. Han, C. T. Chen, L. W. Lo, K. S. Aboody, A. U. Ahmed, and M. S. Lesniak. 2013. Nanoparticleprogrammed self-destructive neural stem cells for glioblastoma targeting and therapy. Small 9 (24): 4123-4129.
[91] Larsen, J. M., D. R. Martin., and M. E. Byrne. 2014. Recent advances in delivery through the blood-brain barrier. Curr. Top. Med. Chem.14 (9): 1148-1160.
[92] Brem, H., M. G. Ewend, S. Piantadosi, J. Greenhoot, P. C. Burger, and M. Sisti. 1995. The safety of interstitial chemotherapy with BCNU-loaded polymer followed by radiation therapy in the treatment of newly diagnosed malignant gliomas: phase I trial. J. Neurooncol. 26:111-123.
[93] Hart, M. G., R. Grant, R. Garside, G. Rogers, M. Somerville, and K. Stein. 2011. Chemotherapy wafers for high grade glioma. Cochrane Database Syst. Rev. 3. doi:10.1002/14651858.CD007294.pub2.
[94] Chowdhary, S. A., T. Ryken, and H. B. Newton. 2015. Survival outcomes and safety of carmustine wafers in the treatment of high-grade gliomas: a meta-analysis. J. Neurooncol. 122 (2): 367-382.
[95] Salahuddin, T. S., B. B. Johansson, H. Kalimo, and Y. Olsson. 1988. Structural changes in the rat brain after carotid infusions of hyperosmolar solutions: a light microscopic and immunohistochemical study. Neuropathol. Appl. Neurobiol. 77: 5-13.
[96] Burgess, A., and K. Hynynen. 2013. Noninvasive and targeted drug delivery to the brain using focused ultrasound ACS Chem. Neurosci.4 (4): 519-526.
[97] Egleton, R. D., and T. P. Davis. 2005. Development of neuropeptide drugs that cross the blood-brain barrier. Neurotherapeutics 2 (1): 44-53.
[98] Ramalho-Santos, M., S. Yoon, Y. Matsuzaki, R. C. Mulligan, and D. A. Melton. 2002. “Sternness”: transcriptional profiling of embryonic and adult stem cells. Science 298 (5593): 597-600.
[99] Sun, J., A. Ramos, B. Chapman, J. B. Johnnidis, L. Le, Y. J. Ho, A. Klein, O. Hofmann, and F. D. Camargo. 2014. Clonal dynamics of native haematopoiesis. Nature 514 (7522): 322-327.
[100] Ning, J., and H. Wakimoto. 2014. Oncolytic herpes simplex virusbased strategies: toward a breakthrough in glioblastoma therapy. Front. Microbiol. 5 (303): 1-13.
[101] Kaufmann, J. K., and E. A. Chiocca. 2014. Glioma virus therapies between bench and bedside. Neuro-Oncology 16 (3): 334-351.
[102] Kaufmann, J. K., and E.A. 2014. Chiocca. Glioma virus therapies between bench and bedside. Neuro-Oncology 16: 334-351.
[103] Russell, S. J., K. W. Peng, and J. C. Bell. 2014. Oncolytic virotherapy. Nat. Biotechnol. 30: 1-29.
[104] Wollmann, G., K. Ozduman, and A. N. van den Pol. 2012. Oncolytic virus therapy of glioblastoma multiforme - concepts and candidates. Cancer J. 18 (1): 69-81.
[105] Rubsam, L. Z., P. D. Boucher, P. J. Murphy, M. KuKuruga, and D. S. Shewach. 1999. Cytotoxicity and accumulation of ganciclovir triphosphate in bystander cells cocultured with herpes simplex virus type 1 thymidine kinase-expressing human glioblastoma cells. Cancer Res. 59: 669-675.
[106] Beck, C., S. Cayeux, S. D. Lupton, B. Dorken, and T. Blankenstein. 1995. The thymidine kinase/ganciclovir-mediated “suicide” effect is variable in different tumor cells. Hum. Gene Ther. 6: 1525-1530.
[107] Colombo, F., L. Barzon, E. Franchin, M. Pacenti, V. Pinna, D. Danieli, M. Zanusso, and G. Palu. 2005. Combined HSV-TK/IL-2 gene therapy in patients with recurrent glioblastoma multiforme: biological and clinical results. Cancer Gene Ther. 12: 835-848.
[108] Perry, J. R. 2012. Thromboembolic disease in patients with high-grade glioma. Neuro-Oncology 14 (Suppl. iv): iv73-iv80.
[109] De Cicco, M. 2004. The prothrombotic state in cancer: pathogenic mechanisms. Crit. Rev. Oncol. Hematol. 50 (3): 187-196.
[110] McKie, E. A., A. R. MacLean, A.D. Lewis, G. Cruickshank, R. Rampling, S.C. Barnett, P.G. Kennedy, S.M. Brown. 1996. Selective in vitro replication of herpes simplex virus type 1 (HSV-1) ICP34.5 null mutants in primary human CNS tumours— evaluation of a potentially effective clinical therapy. Br J Cancer 74 (5): 745-752.
[111] Mineta, T., S. D. Rabkin, T. Yazaki, W. D. Hunter, R. L. Martuza. 1995. Attentuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat. Med. 1 (9): 938-943.
[112] Freeman, A. I., Z. Zakay-Rones, J. M. Gomori, E. Linetsky, L. Rasooly, E. Greenbaum, S. Rozenman-Yair, A. Panet, E. Libson, C.S. Irving, E. Galun, and T. Seigal. 2006. Phase I/II trial of intravenous NDV-HUJ oncolytic virus in recurrent glioblastoma multiforme. Mol. Ther. 13: 221-228.
[113] Rainov, N.G. 2000. A Phase III Clinical evaluation of herpes simplex virus type 1 thymidine kinase and ganciclovir gene therapy as an adjuvant to surgical resection and radiation in adults with previously untreated glioblastoma multiforme. Hum. Gene Ther. 11 (17): 2389-2401.
[114] Yla-Herttuala, M. W. S., J. Martin, P. Warnke, P.Menei, D. Eckland, J. Kinley, R. Kay, and Z. Ram. 2013. Adenovirus-mediated gene therapy with sitimagene ceradenovec followed by intravenous ganciclovir for patients with operable high-grade gliomas (ASPECT): a randomised, open-label, phase 3 trial. Lancet Oncol. 14 (9): 822-833.
[115] Stupp, R., W. P. Mason, M. J. van den Bent, M. Weller, B. Fisher, M.J.B. Taphoorn, K. Belanger, A. A. Brandes, C. Marosi, U. Bogdahn, J. Curschmann, R. C. Janzer, S. K. Ludwin, T. Gorlia, A. Allgeier, D. Lacombe, J. Gregory Cairncross, E. Eisenhauer, and R. O. Mirimanoff. 2005. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Eng. J. Med. 352: 987-996.
[116] Barnett, S. C., L. Robertson, D. Graham, D. Allan, and R. Rampling. 1998. Oligodendrocyte-type-2 astrocyte (O-2A) progenitor cells transformed with c-myc and H-ras form high-grade glioma after stereotactic injection into the rat brain. Carcinogenesis 19 (9): 1529-1537.
[117] O’Neill, D. W., S. Adams, and N. Bhardwaj. 2004. Manipulating dendritic cell biology for the active immunotherapy of cancer. Blood 104 (8): 2235-2246.
[118] Ahmed, M. S., and Y. S. Bae. 2014. Dendritic cell-based therapeutic cancer vaccines: past, present, and future. Clin. Exp. Vaccine 3 (2): 113-116.
[119] Schreiber R. D., L. J. Old, and M. J. Smyth. 2011. Cancer Immunoedit-ing: Integrating immunity’s roles in cancer suppression and promotion. Science 331 (6024): 1565-1570.
[120] Fonteneau, J. F., M. Gilliet, M. Larsson, I. Dasilva, C. Munz, Y. J. Liu, and N. Bhardwaj. 2003. Activation of influenza virus-specific CD4+ and CD8+ T cells: a new role for plasmacytoid dendritic cells in adaptive immunity. Blood 101 (9): 3520-3526.
[121] Salio, M., M. Cella, W. Vermi, F. Facchetti, M. J. Palmowski, C. L. Smith, D. Shepherd, M. Colonna, andV. Cerundulo. 2003. Plasmacytoid dendritic cells prime IFN-gamma-secreting melanoma-specific CD8 lymphocytes and are found in primary melanoma lesions. Eur. J. Immunol. 33: 1052-1062.
[122] Eshhar, Z., T. Waks, G. Cross, and D. G. Schindler. 1993. Specific activation and targeting of cytotoxiclymphocytes through chimeric single chains consisting of antibody-binding domains and the y or C subunits of the immunoglobulinand T-cell receptors. PNAS 90: 720-724.
[123] Miao, H., B. D. Choi, C. M. Suryadevara, L. Sanchez-Perez, S. Yang, G. De Leon, E. J. Sayor, R. McLendon, J. E. Herndon II, P. Healy, G. E. Archer, D. D. Binger, L. A. Johnson, and J. H. Sampson. 2014. EGFRvIII-specific chimeric antigen receptor T cells migrate to and kill tumor deposits infiltrating the brain parenchyma in an invasive xenograft model of glioblastoma. PLos One 9 (4): 1-9.
[124] Lee, D.W., D. M. Barrett, C. Mackall, R. Orentas, and S. A. Grupp. 2012. The future is now: chimeric antigen receptors as new targeted therapies for childhood cancer. CCR Focus 18 (10): 2780-2790.
[125] Imai, C., K. Mihara, M. Andreansky, I. C. Nicholson, C. H. Pui, T. L. Geiger, and D. Campana. 2004. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 18: 676-684.
[126] Carpentino, C., M. C. Milone, R. Hassan, J. C. Simonet, M. Lakhal, M. M. Suhoski, A. Varela-Rohena, K. M. Haines, D. F. Heitjan, S. M. Albelda, R. G. Carroll, J. L. Riley, I. Pastan, and C. H. June. 2009. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. PNAS 106 (9): 3360-3365.
[127] Zhao, Y., Q. J. Wang, S. Yang, J. N. Kochenderfer, Z. Zheng, X. Zhong, M. Sadelain, Z. Eshhar, S. A. Rosenberg, and R. A. Morgan. 2009. A herceptin-based chimeric antigen receptor with modified signaling domains leads to enhanced survival of transduced T lymphocytes and antitumor activity. J. Immunol. 183 (9): 5563-5574.
[128] Morgan, R. A. 2013. Risky business: target choice in adoptive cell therapy. Blood 122 (20): 3392-3394.
[129] Brentjens, R., R. Yeh, Y. Bernal, I. Riviere, and M. Sadelain. 2010. Treatment of chronic lymphocytic leukemia with genetically targeted autologous T cells: case report of an unforeseen adverse event in a phase I clinical trial. Mol. Ther. 18 (4): 666-668.
[130] Kochenderfer, J. N., M. E. Dudley, S. A. Feldman, W. H. Wilson, D. E. Spaner, I. Maric, M. Stetler-Stevenson, G. Q. Phan, M. S. Hughes, R. M. Sherry, J. C. Yang, U. S. Kammula, L. Devillier, R. Carpenter, D. A. Nathan, R. A. Morgan, C. Laurencot, and S. A. Rosenberg. 2012. B-cell depletion and remissions of malignancy along with cytokineassociated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 119 (12): 2709-2720.
[131] Grupp, S. A., M. Kalos, D. Barrett, R. Aplenc, D. L. Porter, S. R. Rheingold, D. T. Teachey, A. Chew, B. Hauck, J. Fraser Wright, M. C. Milone, B. L. Levine, and C. H. June. 2013. chimeric antigen receptor-modified t cells for acute lymphoid leukemia. N Engl. J. Med. 368 (16): 1509-1518.
[132] Le Huu, D., T. Matsushita, G. Jin, Y. Hamaguchi, M. Hasegawa, K. Takehara, and M. Fujimoto. 2012. IL-6 blockade attenuates the development of murine sclerodermatous chronic graft-versus-host disease. J. Invest. Dermatol. 132: 2752-2761.
[133] Pule, M. A., B. Savoldo, G. D. Myers, C. Rossig, H. V. Russell, G. Dotti, M. H. Huls, E. Liu, A. P. Gee, Z. Mei, E. Yvon, H. L. Weiss, H. Liu, C. M. Rooney, H. E. Heslop, and M. K. Brenner. 2008. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat. Med. 14 (11): 1264-1270.
[134] Hatiboglu, M. A., J. Wei, A. S. G. Wu, and A. B. Heimberger. 2010. Immune therapeutic targeting of glioma cancer stem cells. Target Oncol. 5 (3): 217-227.
[135] Barkholt, L., E. Flory, V. Jekerle, S. Lucas-Samuel, P. Ahnert, L. Bisset, D. Büscher, W. Fibbe, A. Foussat, M. Kwa, O. Lantz, R. Mačiulaitis, T. Palomäki, C. K. Schneider, L. Sensebé, G. Tachdjian, K. Tarte, L. Tosca, and P. Salmikangas. 2013. Risk of tumorigenicity in mesenchymal stromal cell-based therapies-bridging scientific observations and regulatory viewpoints. Cytotherapy 15 (7): 753-759.
[136] Harsh, G. R., T. S. Deisboeck, D. N. Loius, J. Hilton, M. Colvin, J. S. Silver, N. H. Qureshi, J. Kracher, D. Finkelstein, E. A. Chiocca, and F. H. Hochberg. 2000. Thymidine kinase activation of ganciclovir in recurrent malignant gliomas: a gene-marking and neuropathological study. J. Neurosurg. 92 (5): 804-811.
[137] Smith, A.G. 2001. Embryo-derived stem cells: of mice and men. Annu. Rev. Cell Dev. Biol. 17: 435-462.
[138] Le Blanc, K., and M. F. Pittenger. 2005. Mesenchymal stem cells: progress toward promise. Cytotherapy 7 (1): 36-45.
[139] Tolar, J., A. J. Nauta, M. J. Osborn, A. P. Mortari, R. T. McElmurry, S. Bell, L. Xia, N. Zhou, M. Riddle, T. M. Schroeder, J. J. Westendorf, R. S. McIvor, P. C. W. Hogendoorn, K. Szuhai, L. Oseth, B. Hirsch, S. R. Yant, M. A. Kay, A. Peister, D. J. Prockop, W. E. Fibbe, and B. R. Blazar. 2007. Sarcoma derived from cultured mesenchymal stem cells. Stem Cells 25: 371-379.
[140] Atsma D. E., W. E. Fibbe, and T. J. Rabelink. 2007. Opportunities and challenges for mesenchymal stem cell-mediated heart repair. Curr. Opin. Lipidol. 18 (6): 645-649.
[141] Solchaga, L. A., K. J. Penick, and J. F. Welter. 2011. Chondrogenic differentiation of bone marrow-derived mesenchymal stem cells: Tips and Tricks. Methods Mol. Biol. 698: 253-278.
[142] Wei X., X. Yang, Z. P. Han, F. F. Qu, L. Shao, and Y. F. Shi. 2013. Mesenchymal stem cells: a new trend for cell therapy. Acta Pharmacol. Sinica 34 (6): 747-754.
[143] Ryan, J. M., F. P. Barry, J. M. Murphy, and B. P. Mahon. 2005. Mesenchymal stem cells avoid allogeneic rejection. J. Inflam. 2:8.
[144] Medvedev, S. P., A. I. Shevchenk, and S. M. Zakian. 2010. Induced pluripotent stem cells: problems and advantages when applying them in regenerative medicine. ACTA NAT. 2 (5): 18-28.
[145] Yamanaka, S. 2009. A FRESH Look at iPS cells. Cell 137 (1): 13-17.
[146] Chan, T. M., J. Y. R. Chen, L. I. Ho, H. P. Lin, K. W. Hsueh, D. D. Liu, Y. H. Chen, A. C. Hsieh, N. M. Tsai, D. Y. Hueng, S. T. Tsai, P. W. Chou, S. Z. Lin, and H. J. Harn. 2014. ADSC Therapy in neurodegenerative disorders. Cell Transplant. 23 (4-5): 549-557.
[147] Desplats P., H. J. Lee, E. J. Bae, C. Patrick, E. Rockenstein, L. Crews, B. Spencer, E. Masliah, and S. J. Lee. 2009. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alphasynuclein. PNAS 106 (31): 13010-13015.
[148] Sanberg, P. R., D. J. Eve, A. E. Willing, S. Garbuzova-Davis, J. Tan, C. D. Sanberg, J. G. Allickson, E. L. Cruz, and C. V. Borlongan. 2011. The treatment of neurodegenerative disorders using umbilical cord blood and menstrual blood-derived stem cells. Cell Transplant. 20 (10): 85-94.
[149] Spinelli V., P. V. Guillot, and P. De Coppi. 2013. Induced pluripotent stem (iPS) cells from human fetal stem cells (hFSCs). Organogenesis 9(2): 101-110.
[150] Broxmeyer, H. E. 2010. Umbilical cord transplantation: Epilogue. Semin. Hematol. 47 (1): 97-103.
[151] Altaner, C., V. Altanerova, M. Cihova, K. Ondicova, B. Rychly, L. Baciak, and B. Mravec. 2014. Complete regression of glioblastoma by mesenchymal stem cells mediated prodrug gene therapy simulating clinical therapeutic scenario. International J. Cancer 134 (6): 1458-1465.
[152] Egleton, R. D., and T. P. Davis. 2005. Development of neuropeptide drugs that cross the blood-brain barrier. NeuroRx 2 (1): 44-53.
[153] Qin, J., X. Yang, J. Mi, J. Wang, J. Hou, T. Shen, Y. Li, B. Wang, X. Li, and W. Zhu. 2014. Enhanced antidepressant-like effects of the macromolecule trefoil factor 3 by loading into negatively charged liposomes. Int. J. Nanomed. 9: 5247-5257.
[154] Wang, X., P. Liu, W. Yang, L. Li, P. Li, Z. Liu, Z. Zhuo, and Y. Gao. Microbubbles coupled to methotrexate-loaded liposomes for ultrasound-mediated delivery of methotrexate across the blood-brain barrier. Int. J. Med.; 9: 4899-4909.
[155] Steiniger, S. C., J. Kreuter, A. S. Khalansky, I. N. Skidan, A. I. Bobruskin, Z. S. Smirnova, S. E. Severin, R. Uhl, M. Kock, K. D. Geiger, and S. E. Gelperina. Chemotherapy of glioblastoma in rats using doxorubicin-loaded nanoparticles. Int. J. Cancer. 109 (5): 759-767.
[156] Jones, A. R., and E. V. Shusta. 2007. Blood-Brain Barrier Transport of Therapeutics via Receptor-Mediation. Pharm. Res. 24(9): 1759-1771.
[157] Pardridge, W. M. 2001. Brain Drug Targeting and Gene Technologies. Jpn. J. Pharmacol. 87 (2): 97-103.
[158] Wait, S. D., R. S. Prabhu, S. H. Burri, T. G. Atkins, and A. L. Asher. 2015. Polymeric drug delivery for the treatment of glioblastoma. Neuro-Oncology 17 (2): ii9-ii23.
[159] Larsen, J.M., D. R. Martin, and M. E. Byrne. 2014. Recent advances in delivery through the blood-brain barrier. Curr. Top. Med. Chem. 14 (9): 1148-1160.
[160] Jewell, C.M., S. C. Bustamante Lopez, and D. J. Irvine. 2011. In situ engineering of the lymph node microenvironment via intranodal injection of adjuvant-releasing polymer particles. PNAS 108 (38): 15745-15750.
[161] Burgess, A., C.A. Ayala-Grosso, M. Ganguly, J.F. Jordao, I. Aubert, K. Hynynen. 2011. Targeted Delivery of Neural Stem Cells to the Brain Using MRI-Guided Focused Ultrasound to Disrupt the Blood-Brain Barrier. PLoS ONE 6 (11): e27877.
[162] Balyasnikova, I. V., M. S. Prasol, S. D. Ferguson, Y. Han, A. U. Ahmed, M. Gutova, A. L. Tobias, D. Mustafi, E. Rincon, L. Zhang, K. S. Aboody, and M. S. Lesniak. 2014. Intranasal delivery of mesenchymal stem cells significantly extends survival of irradiated mice with experimental brain tumors. Mol. Ther. 22: 140-148.
[163] Sampson, J. H., K. D. Alpade, G. E. Archer, A. Coan, A. Desjardins, A. H. Friedman, H. S. Friedman, M. R. Gilbert, J. E. Herndon, R. E. McLendon, D. A. Mitchell, D. A. Reardon, R. Sawaya, R. Schmittling, W. Shi, J. J. Vredenburgh, D. D. Bigner, and A. B. Heimberger. 2011. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro-Oncology 13 (3): 324-333.
[164] Phuphanich, S., C. J. Wheeler, J. D. Rudnick, M. Mazer, H. Q. Wang, M. A. Nuno, J. E. Richardson, X. Fan, J. Ji, R.M. Chu, J. G. Bender, E. S. Hawkins, C. G. Patil, K. L. Black, and J. S. Yu. 2013. Phase I trial of a multi-epitope-pulsed dendritic cell vaccine for patients with newly diagnosed glioblastoma. Cancer Immunol. Immunother. 62: 125-135. [165] Fadul, C. E., J. L. Fisher, T. H. Hampton, E. C. Lallana, Z. Li, J. Gui, Z. M. Szczepiorkowki, T. D. Tosteson, C. H. Rhodes, H. A. Wishart, L. D. Lewis, and M. S. Ernstoff. 2011. Immune response in patients with newly diagnosed glioblastoma multiforme treated with intranodal autologous tumor lysate-dendritic cell vaccination after radiation chemotherapy. J. Immunother. 34 (4): 382-389.
[166] Shand, N., F. Weber, L. Mariani, M. Bernstein, A. Gianella-Borradi, Z. Long, A. G. Sorensen, and N. Barbier. 1999. A phase 1-2 clinical trial of gene therapy for recurrent glioblastoma multiforme by tumor transduction with the herpes simplex thymidine kinase gene followed by ganciclovir. Hum. Gene Ther. 10 (14): 2325-2335.
[167] Markert, J. M., P. G. Liechty, W. Wang, S. Gaston, E. Braz, M. Karrasch, L. B. Nabors, M. Markiewicz, A. D. Lakeman, C. A. Palmer, J. N. Parker, R. J. Whitley, G. Y. Gillespie. 2009. Phase Ib trial of mutant herpes simplex virus G207 inoculated pre-and post-tumor resection for recurrent GBM. Mol. Ther. 17 (1): 199-207.
International Journal of Translational Science, Vol. 1, 67–106.
doi: 10.13052/ijts2246-8765.2015.005
© 2015 River Publishers. All rights reserved.